PacBio Iso-Seq Analysis
With long and accurate HiFi reads, you can characterize the full diversity of the transcriptome
Using the long reads generated by Single Molecule, Real-Time (SMRT) sequencing PacBio Iso-Seq method provides reads that span entire transcript isoforms, eliminating the need for computational reconstruction, and enabling functional characterization of full-length transcript isoforms. The analysis is performed de novo, without a reference genome. The Iso-Seq application provides accurate information about alternatively spliced exons and transcriptional start and end sites.

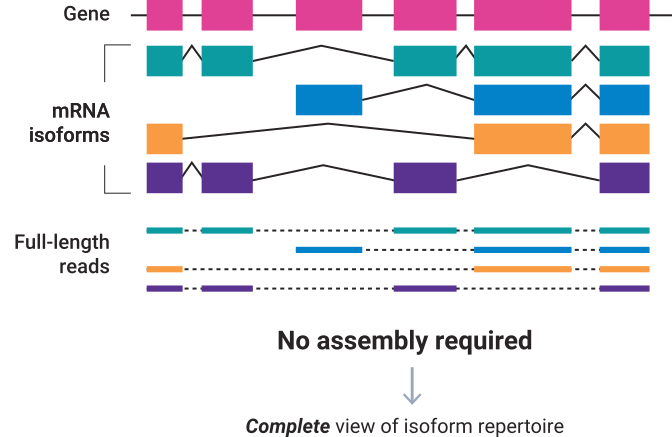
Iso-Seq method can be used to:
- Profile whole transcriptomes exhaustively at the isoform level
- Characterize alternative start and polyadenylation sites as well as exon skipping events
- Detect isoforms as potential biomarkers
- Target genes of interest using synthetic probes with sample multiplexing to query isoforms in a cost-effective manner
- Discover novel genes and informs even in well-characterized model organisms
- Create a reference transcriptome for better RNA-Seq quantification at isoform-level resolution
- Generate full-length transcript isoforms that can be confidently assigned to individual cells
- Move beyond 3’ gene counting to include TSS, polyA, and complete exon connectivity data to deepen your understanding of cell type differences
- Distinguish cell types that play unique roles in complex systems like the immune system
- Understand how alternative splicing of critical genes drives function in tissues like the brain
Full-length, HiFi reads (Q20, single-molecule resolution) are identified and clustered at the isoform level, then polished to create high-quality consensus.
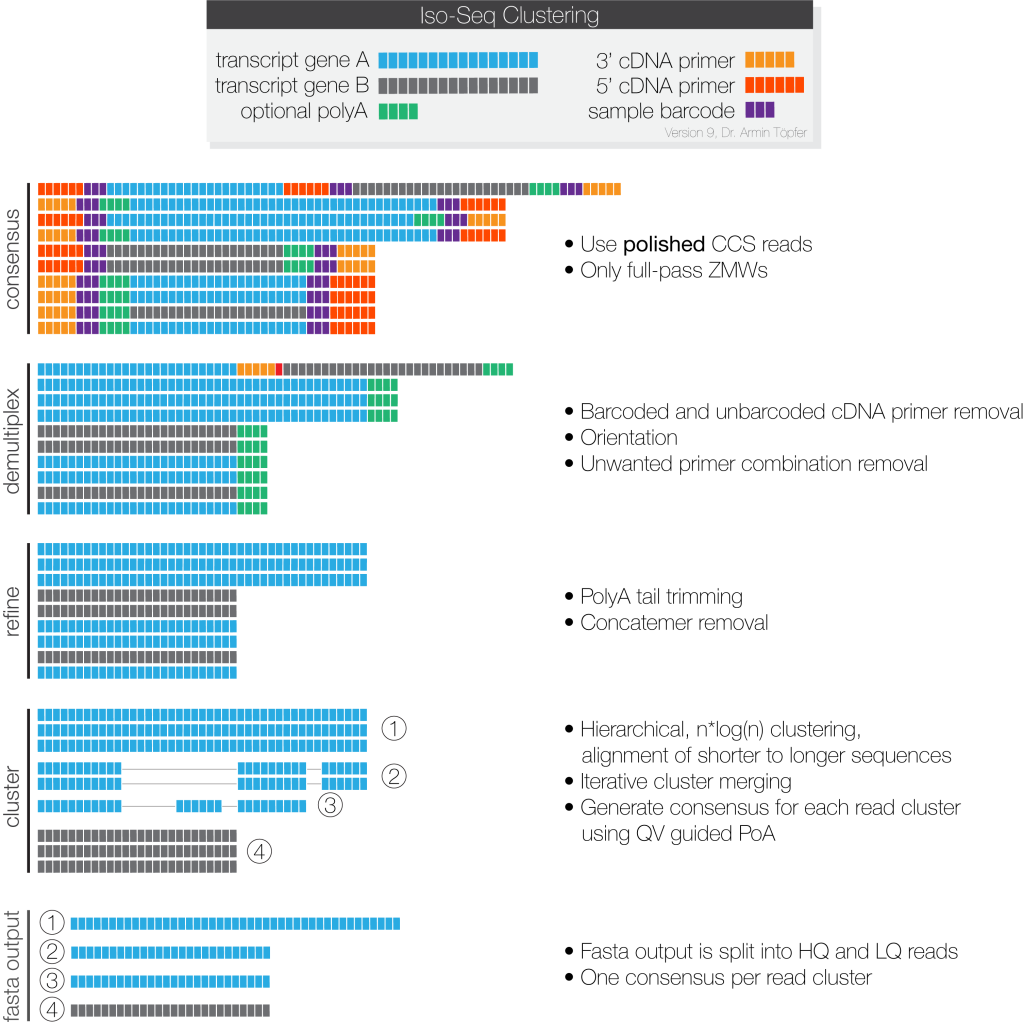
NGI provides following analysis results:
- Full-Length Non-Concatemer Read Assignments: Full-length reads that have primers and polyA tails removed, in BAM format.
- Full-Length Non-Concatemer Report: Includes strand, 5’ primer length, 3’ primer length, polyA tail length, insertion length, and primer IDs for each full-length read that has primers and polyA tail, in CSV format.
- High-Quality Isoforms: Isoforms with high consensus accuracy, in FASTQ and FASTA format. This is the recommended output file to work with.
- Low-Quality Isoforms: Isoforms with low consensus accuracy, in FASTQ and FASTA format. We recommend that you work only with High-Quality isoforms, unless there are specific reasons to analyze Low-Quality isoforms.
- Full-length Non-Concatemer Read Assignments: Report of full-length read association with collapsed filtered isoforms, in text format.
- Collapsed Filtered Isoform Counts: Report of read count information for each collapsed filtered isoform.
If reference is provide, additional results include:
- Mapped High Quality Isoforms: Alignments mapping isoforms to the reference genome, in BAM and BAI (index) formats.
- Collapsed Filtered Isoforms GFF: Mapped, unique isoforms, in GFF format. This is the Mapping step output that is the recommended output file to work with.
- Collapsed Filtered Isoforms: Mapped, unique isoforms, in FASTQ format. This is the Mapping step output that is recommended output file to work with.
- Collapsed Filtered Isoforms Groups: Report of isoforms mapped into collapsed filtered isoforms.
Last Updated: 11th September 2024