De novo sequencing
This method is useful to build novel reference genomes, which could serve as a foundation for future research. Long-read technologies such as PacBio and ONT can decipher much of the structural properties of a genome. While PacBio HiFi assemblies do not need to be polished with short reads, Nanopore data requires an extra polishing step using Illumina data, e.g. paired-end libraries or HiC. HiC adds an additional layer of information to long-read data, arranges scaffolds in chromosomes and proofreads assembly quality.
NGI now offers de novo projects as one single package. Users can send in their sample(s) and NGI will take care of the separate library preparation setups suited for your particular project. A typical setup involves an initial draft genome assembled from long sequence reads, followed by scaffolding to get longer contigs and error-correction. This is followed by annotation of the new reference genome, eg. of genes and other functional elements. We also offer DNA extraction as a service for de-novo projects if required. For more info, please refer to our recent online webinar.
Project setup
In order to know how contiguous your assembly should be, please have a look at the flowchart.
Each study setup is described in more detail below. Once you have chosen the setup suitable for your de-novo project, the arrows direct you to the type of data you need. You can read more about the different technologies NGI offers to generate the data in the technology section below.
More info about what applications, methods and bioinformatics options NGI provides can be found further down.
All new projects should first be discussed with us prior to applications. Please contact us here.
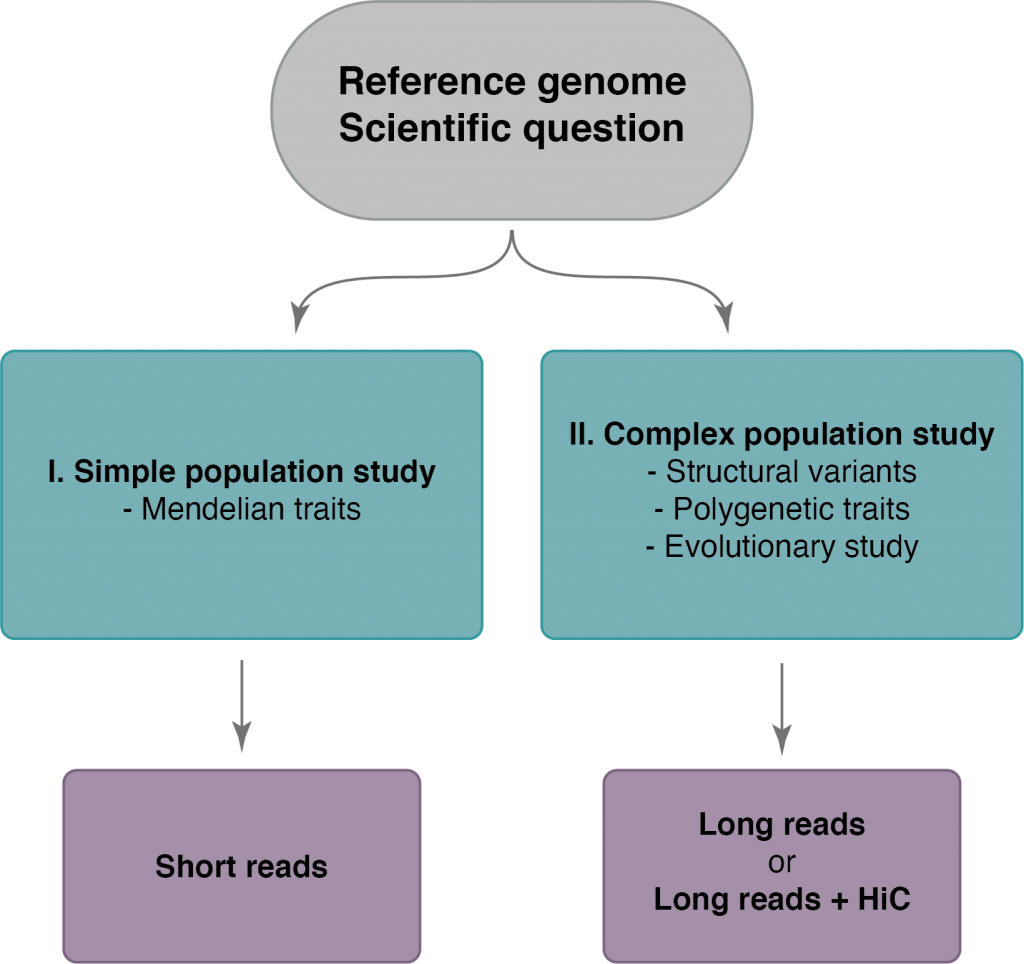
I. Simple population study
Applications
- Genome-wide association studies (GWAS)
- Marker-assisted breeding
- Studies involving Mendelian traits, etc.
They are commonly used to:
- Find and select signals (predominantly SNPs)
- Study allelic frequencies between populations, limited to small blocks of genomic regions.
These assemblies result from scaffolded paired-end Illumina reads and have:
- A high base quality
- Very low contiguity (N50 in kb-range)
- A sole possibility for algorithm-based haplotyping
Structural variation analysis is very cumbersome, and mainly short indels can be analysed. It can be problematic to predict if the observed variation is present at a single locus, or is a part of a larger genomic structure. Genomic repeats are usually collapsed, or mis-assembled; gene duplication events can be problematic to detect.
II. Complex population study
Long-read only assemblies (PacBio or Nanopore)
- The latest iteration of PacBio chemistry (HiFi) does not require additional polishing.
- Nanopore data should be polished by Illumina PE-reads.
- Assemblies exhibit dramatic increases in contiguity – megabase-size contigs (not scaffolds), and contain fewer gaps.
- Haplotypes can be phased based on long stretches of overlapping sequences.
- Causative differences between alleles can be better detected due to improved contiguity.
- Better handling of short to medium repeat segments.
- Better detection of genome incorporation of retroviruses and transposons.
- Detection and phasing of structural variants of 5-10 kb, even if they are present in multiple copies across the genome. These assemblies are invaluable for studies of complex phenotypes (e.g. polygenic, non-Mendelian traits).
- Complexity posed by evolutionary events can be resolved.
Hybrid long-read and Hi-C assembly
- State-of-the-art level for assembling reference genomes, recognized as standard by EBP.
- Data acquired through long-read sequencing that are polished/scaffolded with, and arranged into chromosomes by HiC.
- The highest possible contiguity and base quality are achieved, and assemblies with one contig per chromosome are often observed with this method.
- Recommended by reference genome sequencing initiatives as it yields data with higher contiguity than for long-read assemblies alone. This is essential for studies on complex genomic regions (e.g. MHC), centromeric and telomeric repeats, and on previously unknown parts of genomes of functional interest (“genomic dark matter”).
Technologies offered at NGI
- PacBio Revio
Long-read technology which enables contig assembly.
– Guidelines for sample preparation done by users
– HMW DNA extraction as a service
– Assembly strategies – contact NGI
– Sequencing and analysis costs for long-read sequencing
- Oxford Nanopore (ONT) MinION and PromethION
Long-read technology which enables contig assembly.
– Guidelines for sample preparation done by users
– HMW DNA extraction as a service
– Assembly strategies – contact NGI
– Sequencing and analysis costs for long-read sequencing
- Illumina for scaffolding purposes – HiC or Omni-C
HiC and Omni-C are a powerful scaffolding tools that are also useful for polishing, SV Detection, SNP Calling and Phasing.
– Sample requirements – Arima HiC or Dovetail Omni-C
– Illumina library prep and sequencing costs
- Illumina for polishing purposes
Illumina short-read data is used for lower-quality scaffold assemblies, as well as for polishing long-read data.
– Sample requirements – Illumina TruSeq DNA PCR-free
– Illumina library prep and sequencing costs
- Other valuable technologies for de-novo project but are currently not available at NGI:
– BioNano optical maps
– Linkage maps
Assembly
Methods for the initial sequencing of genomic DNA in order to build a draft genome reference.Scaffolding
Methods to scaffold contigs together and correct genome assembly errors.Annotation
RNA sequencing methods that can be used to annotate de-novo genomes with transcript locations.Illumina TruSeq Stranded mRNA
RNA sequencing of mRNAs selected through poly-A enrichment.
truseq mRNA illumina RNA-Seq library preparation transcriptomics RNADovetail Omni-C
A proximity-ligation protocol using a sequence-independent endonuclease, generating data for TAD identification and scaffolding.
chromatin scaffolding library preparation epigenetics TADs illumina de novoG‑SPLAT: Genomic SPLAT for Whole Genome Sequencing
G-SPLAT is a library preparation technique designed for low-quality or limited-input DNA, with applications in whole-genome sequencing, metagenomics, and and other challenging sequencing projects.
genome illumina WGS dna single-stranded DNA ssDNA library preparationIllumina DNA
Low cost library preparation option for gDNA based on bead-linked transposase. Only for full plates of samples.
normalization library preparation genome illumina WGS dna nexteraIllumina DNA PCR-Free
Method for shotgun DNA libraries used for whole genome sequencing and metagenomics.
illumina WGS dna tagmentation PCR-free library preparation genomeIllumina TruSeq DNA PCR-free
Gold standard method for shotgun DNA libraries used for whole genome sequencing and metagenomics.
genome illumina WGS dna library preparation truseqIllumina TruSeq DNA Nano
Library preparation from limited input DNA, used in whole genome sequencing and metagenomics etc.
library preparation truseq genome illumina WGS dnaThruPLEX DNA-seq
Library preparation for DNA, ideal for preparing libraries from small amounts of input material. Works well for shotgun libraries, ChIP DNA and FFPE samples, amongst others.
library preparation genome illumina WGS dnaNanopore cDNA sequencing
Nanopore cDNA sequencing is able to sequence entire transcripts in one go, ideal for detecting isoforms and fusions events.
assembly long-read nanoporeNanopore DNA sequencing
Nanopore instruments can sequence very long continuous fragments of DNA. Sequencing native DNA allows detection of base modifications.
assembly long-read nanoporeNanopore Direct RNA sequencing
Nanopore direct RNA sequencing is able to sequence entire transcripts from native RNA, opening up opportunities to detect RNA modifications.
long-read nanopore assemblyPacBio SMRT sequencing
PacBio SMRT sequencing generates reads tens of kilobases in length enabling high quality genome assembly, structural variant analysis, amplicon resequencing, full-length transcript isoform sequencing, full-length 16S rRNA sequencing and amplification free epigenetic characterization.
smrt assembly pacbio methylation amplicon hifi de novo iso seq sv revioGenome assemblies with HiFi data
NGI can generate high quality assemblies using IPA and hifiasm assemblers
hifi hifiasm ipa hic omnic revio scaffolding assembly pacbioNanopore QC analysis
Quality control, Basecalling and multiplexing of sequencing reads generated by Oxford Nanopore sequencers.
long-read nanoporePromethION secondary analysis
Additional compute intensive nanopore raw data processing services provided by NGI
methylation base modifications basecalling pod5Short read QC analysis
Basic quality-control monitoring of Illumina FastQ sequence data.
QC fastqc fastq screen checkqc