Bioinformatics
All samples, sequenced at the NGI, undergo a series of quality certified checks. Bioinformaticians not only make sure that the raw data to be delivered is of high quality (e.g., Q30, yield, etc), but also that sequences are biologically relevant by checking the absence of contamination and, depending on the specific application, by checking the results of the primary analysis (e.g. duplicate rates, GC-content, etc).
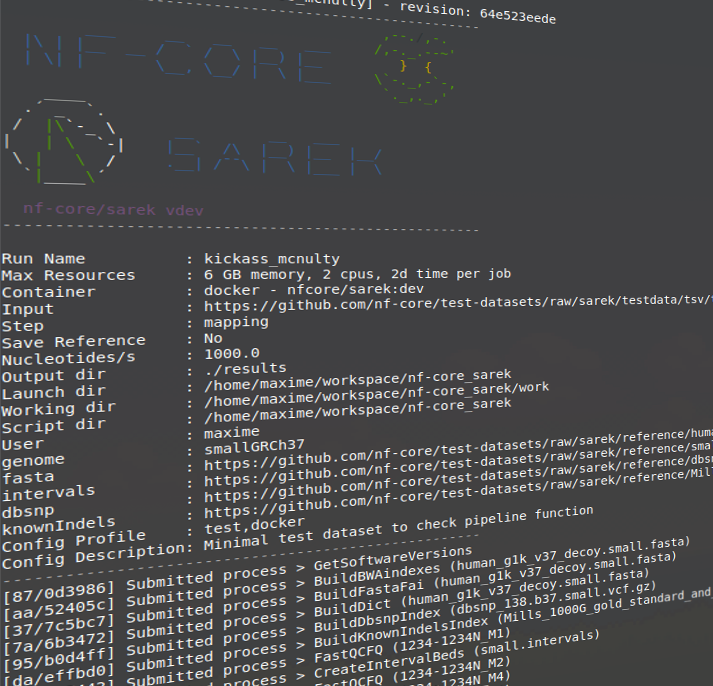
NGI performs automated bioinformatics analyses in a number of applications. Follow the links from different methods to see information about the associated analysis that we perform.
For more extensive help with bioinformatics analysis, we recommend that you contact the National Bioinformatics Infrastructure Sweden (NBIS).