RAD-sequencing
Genotyping-by-sequencing without prior genome information.
Description
RAD (Restriction-site Associated DNA) sequencing is a method by which certain restriction enzymes are used to fragment genomic DNA samples, followed by size selection and sequencing of molecules within a certain size range. This enables reproducible enrichment of a particular set of genomic loci, where the loci retained for sequencing are more or less randomly distributed over the genome. The sequence data resulting from this application may be used for downstream applications, including linkage analysis, inference of population genetics metrics and polymorphism discovery.
Sample requirements
We run RAD-seq library preps in batches of 94 samples. We use two controls per batch, please consult our sample submission page for details on how to load your samples in plates.
| Input | Purified EcoRI-cut DNA. Gel results must be provided. |
| Amount | >200 ng |
| Concentration | 16–100 ng/µL |
| Volume | 16–170 µL. Approx 2 μL of the sample will be used for our initial quality checks, so please account for this when sending us samples. |
| Sample Buffer | 10 mM Tris, pH 8–8.5 or similar (e.g. Qiagen’s EB). |
EcoRI digestion
The EcoRI digestion can be done using EcoRI from any standard supplier, e.g. NEB, in the kit-supplied buffer and using the manufacturer’s suggested protocol (e.g. here for NEB). The recommended protocols will typicallly include a heat-inactivation of the enzyme, followed by an optional cleanup. While we do not require a post-digestion cleanup before sending the samples to NGI, it may be helpful if the samples contain known chemicals/molecules that can inhibit the library preparation.
How to evaluate the sample quality
We check your samples upon arrival, however we still require our users to do their own QC steps before sending samples. For this library prep method, we require the following to be included when submitting sample information.
Making sure the concentration, volume and amount fullfil the criteria listed in the table above (this applies to measurements AFTER restriction digestion and inactivation of the enzyme).
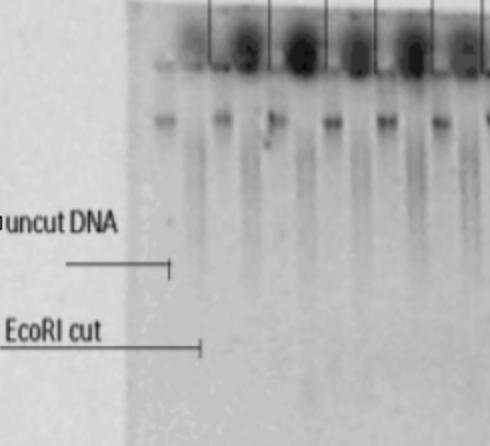
The EcoRI-cut DNA should be run on a gel with digested/undigested samples side-by-side, to confirm successful digestion.
If you are not able to carry out these steps, or your samples are below the required thresholds, please get in touch.
What we do with your samples
Library preparation
During preparation, a custom adapter with complementary bases to the EcoRI overhang is ligated to the ends of the digested DNA using T4 DNA ligase and 10X T4 DNA ligase (New England Biolabs). The ligated adapter contains the partial Illumina adapter. Samples are purified using AMPure XP bead purification (Beckman Coulter), strand displacement is performed using Bst 2.0 polymerase, 10X ThermoPol buffer (New England Biolabs) and dNTPs (Thermo Fisher Scientific). Another bead purification follows before amplification and indexing by PCR. Finally, the libraries are cleaned up in another bead purification.
Library QC and sequencing
To assess the quality of the library, the concentration is measured with the Qubit dsDNA HS assay (Thermo Fisher Scientific) and the fragment size distribution is checked using the Fragment Analyzer (Agilent). A calculated concentration in the range of 420–720 bp is used to even out the different samples’ contribution to the pool before subjecting the pool to size selection with the BluePippin (Sage Science). The concentration and size of the pool is analyzed with Qubit and Fragment Analyzer (or Bioanalyzer) before sequencing according to agreed upon setup.
Expected results
It is expected that the resulting sequence data from each sample is enriched for loci in the genome that were generated by EcoRI cuts and that the libraries from each sample will feature mostly the same sequenced loci.
Bioinformatics
NGI can run a no-guarantees preliminary analysis with STACKS: http://catchenlab.life.illinois.edu/stacks/ at no extra cost. This will give a list of the identified loci, and serve as a starting point for comparing polymorphic loci between individuals. This analysis does not require a reference genome.
Price examples
Library preparation cost is ca 32000 SEK per plate (max 94 samples). Typically not much sequencing is required, several plates (maximum 4 plates can be multiplexed) can be sequenced in 1 unit of sequencing (ca 180 GBp).
Last Updated: 9th June 2026